




CONTRIBUTION à l'étude de la fixation de l'azote sous forme d'oxyde au moyen de l'effluve
THESE PRÉSENTÉ A LA FACULTÉ DES SCIENCES DE L'UNIVERSITÉ DE GENÈVE POUR L'OBTENTION DU GRADE DE DOCTEUR ÈS SCIENCES PHYSIQUES
de Genève Ingénieur-chimiste diplômé de l'Université de Genève Assistant au Laboratoire de Chimie théorique et technique de l'Université de Genève
GENÈVE IMPRIMERIE JENT, S. A., RUE NECKER, 9-11 Maison attitrée à la Société Coopérative d'impression 1926
Thèse No 790.
UNIVERSITÉ DE GENÈVE Faculté des Sciences
La Faculté des Sciences, sur le préavis de M. le Prof. E. Briner, autorise l'impression de la thèse présentée par M. Ch. Boissonnas, Ing.-Chim., intitulée: «Contribution à l'étude de la fixation de l'azote sous forme d'oxyde au moyen de l'effluve», sans exprimer d'opinion sur les propositions qui y sont énoncées.
Genève, le 19 janvier 1926. Le Doyen L. W. COLLET
A MONSIEUR LE PROFESSEUR E. BRINER
Ces recherches ont été effectuées dans les laboratoires de chimie théorique et technique de l'Université de Genève, sous la direction de Monsieur le Professeur E. Briner, auquel je me permets d'exprimer ici toute ma reconnaissance.
INTRODUCTION
L'utilisation des décharges électriques pour fixer l'azote sous forme d'oxydes, d'ammoniac, ou d'acide cyanhydrique, a fait l'objet de très nombreuses recherches. Malgré celà, le mécanisme de cette fixation est loin d'être élucidé, en raison surtout de la complexité extrême des phénomènes. auxquels prennent part à la fois des actions thermiques, électroniques et photochimiques 1. C'est cette complexité qui explique en particulier que le champ d'investigation relatif à ce domaine, au lieu de se circonscrire, s'élargit constamment et offre de nouveaux aperçus qu'il convient d'examiner. L'une de ces directions nouvelles nous a paru tout spécialement digne d'être prise en considération en vue de tenter d'améliorer le rendement de la fixation de l'azote sous forme d'oxydes d'azote. Ce rendement, ainsi qu'on l'a relevé à plusieurs reprises, est en effet très faible, puisqu'au moyen de l'arc, qui est actuellement le meilleur moyen de fixation sous forme d'oxydes, à peine 3 % de l'énergie consommée se retrouve dans l'oxyde d'azote formé. Pour élever ce rendement, on est limité d'une part par la rétrogradation que subit l'oxyde d'azote lors du refroidissement, d'autre part, par le maximum de concentration atteint par tous les composés endothermiques lorsqu'on élève la température 2. Il nous a semblé qu'il y aurait une voie intéressante à suivre en opérant avec des décharges beaucoup plus froides que l'arc, et en se servant d'un
appareillage permettant une participation plus grande des électrodes au phénomène.
Des recherches antérieures sur la formation de l'ammoniac 1 et sur celle de l'oxyde d'azote 2 ont en effet montré que le matériel des électrodes pouvait exercer une action favorable. C'est pour cette raison que, dans les recherches qui font l'objet de ce travail, nous nous sommes servi de l'effluve électrique 3. Cette décharge, par le fait qu'elle est répartie dans un grand volume, ne donne pas lieu à des élévations de température très marquées; d'autre part, la nature des électrodes, de grande surface, doit avoir une action beaucoup plus importante que dans l'arc.
Des recherches dans cette direction ont été déjà entreprises à la température ordinaire dans les Laboratoires de chimie technique et théorique de Genève 4, mais les résultats obtenus n'ont pas été bien caractéristiques. Il était donc indiqué de reprendre des recherches en opérant à des températures élevées, de manière à intensifier encore davantage les actions catalytiques exercées par le matériel des armatures.
C'est pour réaliser ces conditions de travail que nous avons été amenés à établir un appareillage approprié, qui sera décrit plus loin. Nous avons étudié l'action d'armatures de platine, dont on connaît l'intense activité catalytique, puis nous avons fait quelques essais avec un mélange d'oxydes alcalino-terreux. Cette dernière armature a été choisie en raison de ses propriétés émissives électroniques particulièrement marquées, lesquelles suivant certains résultats trouvés récemment 5, favoriseraient, dans certaines régions de température, la fixation de l'azote sous forme d'oxydes.
Comme l'effluve jaillissant dans le système azoteoxygène donne lieu à la production d'ozone, nous avons dû en même temps prendre. en considération cette production. A ce point de vue, il y avait lieu d'examiner dans quelle mesure l'ozone pouvait être capable, par lui-même, de provoquer l'oxydation de l'azote. Or, ainsi qu'on le verra, cette oxydation ne se produit pas, tout au moins d'une façon appréciable, même lorsque l'azote est mis en présence de l'ozone à des pressions, c'est-à-dire à des concentrations élevées.
Les méthodes de travail et les résultats obtenus
seront exposés dans les chapitres suivants, dont le
premier sera consacré à la description d'un nouveau
procédé d'analyse de l'oxyde d'azote.. Etant donné les
faibles quantités d'oxydes formés, il nous fallait, en
effet, une méthode à la fois sensible et précise.
I. MÉTHODE D'ANALYSE DES OXYDES D'AZOTE
Nous avons été amenés à perfectionner la méthode DEVARDA, consistant à réduire les nitrates et nitrites en ammoniac que l'on distille dans une solution titrée d'acide sulfurique. Telle qu'on l'utilise couramment, cette méthode est, en effet, insuffisamment précise pour doser de petites quantités d'oxydes d'azote.
Le gaz contenant les oxydes d'azote en faible concentration était laissé plusieurs heures en présence d'une solution de potasse à 30% dans un grand ballon de verre, ce qui permettait une absorption complète. Les petites quantités d'ammoniac, formées par réduction au moyen de l'alliage DEVARDA, étaient reçues clans une solution d'acide sulfurique dilué. Quelques essais nous ont montré qu'il n'est pas possible de titrer des solutions diluées en observant la neutralité au moyen d'indicateurs colorés, la sensibilité de ces derniers étant alors nettement insuffisante. C'est pourquoi nous avons cherché à utiliser la méthode conductométrique déjà préconisée par DUTOIT et DUBOUX pour le dosage de l'ammoniac dans les vins. Lors de la titration d'un acide par une base, les courbes représentant la conductibilité en fonction de la quantité de réactif ajouté, sont formées par deux droites dont l'intersection correspond à la neutralité. Nous nous sommes servi de la cuve classique. Afin d'éviter, au cours d'une titration, les variations de conductibilité provenant de la variation de la concentration due à l'addition du réactif, nous avons titré, ainsi qu'on le fait généralement, une solution diluée d'acide par une solution relativement concentrée de potasse.
L'appareil à distiller était tout d'abord constitué par une colonne ascendante en verre, garnie de perles de verre sur une longueur de 15 cm., suivie d'un réfrigérant également en verre. Nous avons procédé à une série d'essais à blanc, qui ont montré un important passage de produits alcalins (correspondant à 0, 560 cm3 de potasse N-50), variable dans une large mesure suivant les essais, malgré le soin apporté à opérer dans des conditions toujours identiques.
Les causes de ce passage variable de produits alcalins ont été. déterminées par un grand nombre d'essais à blanc. Ce sont les suivantes:
1° Solubilité du verre dans la vapeur d'eau. C'est la cause principale de cette alcalinité. Le remplacement de l'appareil de verre par un appareil de quartz n'a fait qu'augmenter les difficultés, la vapeur d'eau attaquant le quartz et ajoutant au distillat une acidité qui contribuait encore à l'instabilité des essais à blanc. Un appareil construit entièrement en étain nous a, par contre, donné entière satisfaction. Le passage d'alcalinité se trouvait de ce fait diminué de moitié.
2° Alcalinité due à l'eau distillée. L'eau distillée utilisée dans les laboratoires de l'Ecole de Chimie, contient des composés azotés réductibles en ammoniac (concentration N-20.000). Elle est soumise à une purification par ébullition en présence d'alliage DEVARDA en solution légèrement alcaline.
3° Alcalinité due à la potasse. Cette alcalinité est variable dans une large mesure avec la qualité de potasse utilisée. Elle est éliminée par ébullition prolongée de la solution avec un peu d'alliage DEVARDA, jusqu'à disparition de la réaction Nessler.
4° Alcalinité due à l'alliage DEVARDA. Cette alcalinité due probablement à la présence d'azoture d'aluminium dans l'alliage, peut être éliminée par ébullition prolongée de l'alliage finement pulvérisé avec de l'eau distillée. Mais il perd une partie de ses propriétés réductrices; la
cause d'erreur apportée par l'alliage étant sensiblement constante, on peut, sans grand inconvénient, supprimer cette purification.
En plus de la purification des produits, suivant les indications précédentes, il faut observer certaines précautions. Il faut éviter notamment le séjour prolongé de la potasse titrée dans les burettes, à cause de l'enrichissement en alcali par attaque du verre, la surface de contact entre le verre et la solution étant grande, comparée au volume. On ne saurait aussi prendre trop de précautions pour opérer dans des conditions toujours constantes.
Nous avons ainsi obtenu des essais à blanc constants à moins de 0,005 cm3 de potasse N-50 près, ce qui nous a permis de doser avec une approximation de 1 % des quantités de NO de l'ordre de 0,2 cm3. La méthode permet donc de mesurer des quantités d'ammoniac à peine décelables au moyen du réactif Nessler.
II. OXYDATION DE L'AZOTE PAR L'OZONE
En 1874, CARIUS 1, mettant en présence de l'ozone et de l'air dans un ballon contenant de l'eau, ne décela aucune formation d'oxydes d'azote. En 1898, SHENSTONE et EVANS 2 cherchent à expliquer la formation d'oxydes d'azote dans l'effluve par l'action de l'azote sur l'ozone. Ils basent leur hypothèse sur le fait que, si le processus d'ozonisation n'est pas suffisamment poussé, aucun peroxyde d'azote n'apparaît. Plus récemment, JAHN 3 remarque que la vitesse de décompoposition
de l'ozone est plus grande de 14% environ en présence d'azote. Il conclut à la vraisemblance d'une oxydation lente de l'azote, probalement suivant la réaction O +N 2 =NO 2 . Enfin, CHAPMAN et JONES 1, reprenant la question de la décomposition de l'ozone, sans citer le travail de JAHN, prétendent que l'azote n'a pas d'effet sur la vitesse de décomposition de l'ozone.
Nous avons pensé qu'il serait intéressant de reprendre cette question.
Deux séries d'expériences ont été effectuées, la première à la pression ordinaire, à des températures variant entre 20° et 200°, la seconde sous pression.
EXPÉRIENCES A LA PRESSION ORDINAIRE.
L'oxygène nécessaire à la préparation de l'ozone est obtenu en chauffant du permanganate de potassium pur dans un tube de verre. Cet oxygène, débarrassé sur de la potasse de l'acide carbonique, est séché sur du pentoxyde de phosphore, puis soumis aux décharges d'un effluveur de verre è deux diélectriques, refroidi dans le mélange alcool-neige carbonique. Le mélange d'ozone et d'oxygène est aspiré. dans un ballon de deux litres, préalablement vidé à la pompe è huile. Lorsque le ballon est rempli aux deux tiers, on introduit de l'azote. Une légère dépression est cependant maintenue, permettant, après plusieurs heures, d'introduire 40 cm3 d'une solution de potasse à 30%, destinée à absorber les oxydes d'azote. L'étanchéité de l'appareil, entièrement construit en verre soudé, est soigneusement vérifiée. Chaque expérience est précédée d'un grand nombre de rinçages à l'oxygène pur, afin de réduire la quantité d'azote contenue dans l'appareil à 5. 10 -5 gr. au maximum, quantité de l'ordre de grandeur de la précision de l'analyse.
FIG. 1Résultats: 1 Press. partielles (mm Hg) Numéro Mél. ozone-oxyg. Azote Durée Cm 3 NO. 80 192 330 24 h. <0,6 81 256 222 23 h. <0,1 116 258 280 23 h. <0,05 121 195 375 48 h. <0,05 134 300 — <0,05
Les quantités d'oxydes d'azote indiquées, sont une limite supérieure. L'oxydation de l'azote par l'ozone est donc nulle ou très faible dans les conditions indiquées.
EXPÉRIENCES SOUS PRESSION.
Pour abaisser cette limite, nous avons augmenté les concentrations dans une large mesure, soumettant le mélange à une forte pression.
Nous avons été amenés à modifier comme suit les dispositifs expérimentaux (voir figure 1). Le mélange ozone oxygène produit dans l'effluveur est liquéfié dans une ampoule de verre, A. L'excès d'oxygène est évaporé, soumis à la décharge et condensé à nouveau afin d'augmenter la concentration en ozone. Répétant plusieurs fois cette opération, on obtient dans l'ampoule de l'ozone presque pur, que l'on distille dans un tube de verre, placé en B, refroidi dans de l'air liquide, jusqu'à remplir celui-ci à moitié. Les petites quantités d'oxydes d'azote qui auraient pu se former dans l'effluve, à partir de traces d'azote provenant de permanganate, restent dans l'ampoule avec la fraction non distillée. On ajoute au mélange liquide un tiers de son
volume d'air liquide frais (contenant 70% d'azote environ). On ferme le tube au chalumeau, puis on le retire de l'air liquide et le porte rapidement dans un tube d'acier à parois épaisses, dans lequel il pénètre à frottement doux. Un bouchon à vis force une rondelle d'étain contre un ressaut circulaire du tube d'acier et en assure la fermeture. Quelques secondes après, le tube de verre se brise à l'intérieur du tube d'acier sous l'influence de la pression. On vérifie l'étanchéité en immergeant le tube dans de l'eau.
Le mélange ozone, oxygène, azote liquides occupe un volume de 1,5 cm 3 environ. Le volume libre à l'intérieur du tube d'acier est de 80 cm 3. La pression à l'intérieur du tube est évaluée à 120 atmosphères approximativement.
Après avoir laissé les deux gaz en présence pendant 24 heures, on plonge le tube d'acier d'ans l'air liquide, on dévisse le bouchon et on fait communiquer le tube avec un ballon de deux litres, vide d'air et dans lequel on a introduit la potasse absorbante. On réchauffe le tube avec une flamme de gaz pour faciliter le dégagement des oxydes d'azote. La dépression créée dans le tube par l'aspiration dans le ballon (dont le volume est le double de celui des gaz), facilite aussi le dégagement. En outre, pour recueillir les composés de l'azote qui auraient pu être retenus à la surface du tube d'acier, on lave celui-ci avec une solution de potasse purifiée. Les gaz sont laissés 24 heures en présence de la potasse pour assurer une absorption parfaite des oxydes.
Résultats:
Ces expériences ont présenté de grandes difficultés. Il arrive fréquemment que le tube de verre contenant les gaz liquéfiés saute avant la fermeture du tube d'acier. D'autre part, des retours de liquide peuvent se produire pendant la distillation, occasionnant de violentes explosions.
Pour ces raisons, un seul essai a donné toute satisfaction.
La quantité d'oxydes d'azote était inférieure à 0,06 cm 3 de NO. Donc, 2 grammes d'ozone en présence de 1 gramme d'azote pendant 24 heures, à la pression de 120 atmosphères, donnent une quantité d'oxydes d'azote inférieure à 0,000,05 grammes de NO. Nous croyons ainsi pouvoir conclure que, dans les conditions ordinaires, l'ozone, même aux concentrations élevées réalisées par la compression, n'est pas capable d'oxyder l'azote, du moins en quantité appréciable. C'est là une raison de ne pas envisager l'ozone comme agent intermédiaire dans l'oxydation de l'azote.
III. FORMATION D'OXYDES D'AZOTE DANS L'EFFLUVE A DES TEMPÉRATURES COMPRISES ENTRE 200° ET 1000°.
Nous ne croyons pas que des recherches aient été entreprises sur la formation d'oxydes d'azotes dans un effluveur soumis, à des températures supérieures à 200°. WARBURG 1 avait cru remarquer qu'à des températures comprises entre 20° et 180°, le rendement en oxydes d'azote diminuait avec celui en ozone lorsque l'on élevait la température.
Nous avons prouvé qu'il n'en est rien et que cette erreur est probablement due au fait que, en présence d'un excès d'ozone, les oxydes d'azote sont très rapidement transformés en N 2 0 5. Si la concentration en ozone diminue au-dessous d'une certaine limite, l'oxydation s'arrête au stade NO 2, corps plus difficilement absorbable, ce qui justifie une diminution apparente du rendement.
FIG. 2Pour obtenir des résultats comparables entre eux, nous nous sommes appliqué à opérer, dans des conditions semblables, aux différentes températures.
L'effluveur le plus souvent utilisé pour les recherches sur la formation des oxydes d'azote est formé de deux tubes concentriques en verre, entre lesquels passent les gaz qu'il s'agit de soumettre à la décharge. Le rôle des parois diélectriques est de répartir la décharge sur une grande surface. Malheureusement, déjà à une température voisine de 200°, le verre ne résiste plus à la décharge. Il est rapidement transpercé et mis ainsi hors d'usage. Nous avons donc dû recourir à un autre matériel. Après de longs tâtonnements, nous avons été amenés à construire un effluveur à deux diélectriques de quartz qui nous a donné satisfaction.
Cet effluveur (voir figure 2) est composé de deux tubes de quartz concentriques, distants de quelques millimètres, entre lesquels on fait circuler l'air. La conductibilité du quartz augmentant très rapidement avec l'élévation de la température, au-dessus de 500°, on obtient facilement un courant de 10 milliampères sous 7.000 ou 8.000 volts, dans un effluveur de dimensions très restreintes 1.
Ce dispositif a l'avantage de permettre l'étude de l'influence de corps tels que le platine, dont on peut recouvrir les parois intérieures de l'effluveur, sans avoir recours aux effluveurs à un diélectrique et une surface métallique, qui donnent lieu à des décharges locales très violentes, ne présentant pas la régularité caractéristique de l'effluve.
L'effluveur est placé dans un four électrique à résistance de nichrome.
Le courant est distribué, à l'extérieur du tube, par une plaque de nickel, et à l'intérieur du tube par de la poudre de charbon. Le tube extérieur se termine du côté de la sortie des gaz par un tube de quartz étroit, dans le but d'éviter un trop long séjour des gaz dans l'effluveur après leur passage dans l'effluve. L'extrémité opposée de l'effluveur, par laquelle les gaz sont introduits, est refroidie par une circulation d'eau qui protège le bouchon de liège noyé dans de la cire à cacheter, assurant la fermeture. La circulation du gaz est assurée par l'aspiration d'une pompe à huile. Le débit est contrôlé par un anémomètre. Des tubes contenant de la potasse, du chlorure de calcium et du pentoxyde de phosphore, sont prévus pour débarrasser le gaz de l'acide carbonique et de l'humidité.
Lorsque la circulation du gaz est établie depuis un certain temps et que la décharge est régulière, on suspend l'action de la pompe et on relie l'appareil à un ballon de 4 litres, vide d'air, qui, se remplissant, crée une aspiration suffisante pour entretenir un courant gazeux de 10 litres-heure sous une pression de 350 mm. pendant 10 minutes. Introduisant de la potasse purifiée dans le ballon, on absorbe les oxydes d'azote qui sont soumis à l'analyse. Dans ces conditions, la récupération des oxydes d'azote est totale. :
Résultats:
Les résultats des expériences, à température élevée sont consignés dans les tableaux et graphiques suivants:
No temper. 1/h press. volts mil. amp. volts amp. gr/volt amp. h gr/kwh.
Electrode platinée 243 1020 10 230 1500 9,5 10 0,73 2,9 244 990 100 730 1800 5,4 10 0,3 1,2 245 645 10 530 2800 10,0 30 0,29 1,16
246 690 10 530 2900 10,5 29 1,56 5,6 247 770 10 530 2400 10,0 24 5,5 22,0 248 615 10 530 1800 10,5 50 1,05 4,2 250 910 10 530 3500 10,0 15 2,05 8,2 252 805 10 530 2700 10,0 37 1,4 5,6 253 885 10 530 3400 25,0 62 1,8 7,2 254 770 10 530 3500 10,0 35 1,4 5,6 255 740 10 530 2900 10,0 30 1,7 6,8
256 770 10 530 2400 10,0 24 2,1 8,4 258 755 10 530 1900 10,0 29 2,0 8,0 259 1030 10 530 2300 10,0 13 2,0 8,0 260 840 10 530 2400 10,0 24 4,3 18,5 261 745 10 530 2800 10,0 28 3,7 14,8 263 787 10 530 2600 10,0 26 2,5 10,0 264 815 10 530 2600 10,0 26 2,3 9,2 265 820 3 530 2500 10,0 25 1,4 5,6 266 820 10 530 2500 10,0 25 2,3 9,2 267 820 20 530 2700 10,0 27 2,95 11,9 268 820 20 650 3400 10,0 34 2,3 9,2 269 825 20 230 2300 10,0 23 2,9 11,8 270 825 10 345 2800 10,0 28 1,8 7,2 271 825 30 345 2800 10,0 28 3,1 12,4
Electrode nue 272 820 10 345 2400 10,0 24 1,9 7,6 273 815 10 345 2700 10,0 27 1,8 7,2 274 760 10 g45 3500 10,0 35 2,0 8,0
Electrode recouverte d'oxydes 275 745 10 345 2950 10,0 30 1,6 6,4 276 835 10 345 2450 10,0 24 5 2,8 11,2 277 930 10 345 1800 10,0 18 2,7 10,8 278 1030 10 345 1450 10,0 14 5 1,9 1,6
La première colonne se rapporte aux numéros d'ordre chronologique des expériences; la seconde contient les températures en degrés centigrades, mesurées au moyen d'un couple thermoélectrique étalonné, placé entre le
tube extérieur de l'effluveur et la paroi du four; la troisième, les débits gazeux en litres par heure; la quatrième, les pressions en millimètres de mercure; la cinquième, la tension aux bornes de l'effluveur, mesurée au moyen d'un voltmètre statique; la sixième, l'intensité de la décharge lue sur un ampèremètre de précision. Les rendements, contenus dans la huitième colonne, ont été évalués, comme dans la plupart des travaux concernant l'action de l'effluve sur l'air atmosphérique en grammes d'HNO 3 par volt-ampère-heure, le produit des volts par les ampères étant indiqué dans la septième colonne. Cependant, nous reportant au travail récent de Warburg 1, donnant au coefficient de puissance du courant dans un effluveur la valeur moyenne de 0,25, nous avons reporté dans la dernière colonne les rendements vrais, en grammes d'HNO 3 par kilowatt-heure. Cette évaluation des rendements, bien qu'elle se rapproche plus de la réalité que celle que l'on emploie généralement, n'est cependant pas tout à fait exacte, le décalage du courant dans un effluveur variant certainement avec la forme et la nature des électrodes.
Ainsi que nous l'avons indiqué précédemment, toutes nos expériences ont été effectuées avec un effluveur de quartz à deux diélectriques. Dans les séries s'étendant des Nos 243 à 245, 246 à 255, et de 256 à 271, le plus petit des deux tubes était recouvert d'une mince couche de platine. Ce dépôt était obtenu en badigeonnant, avec une solution de chlorure de platine que l'on réduisait par chauffage dans la flamme d'un chalumeau. Répétant plusieurs fois cette opération, on obtenait un dépôt suffisamment régulier. Les résultats très faibles de la première série sont dûs probablement à une mauvaise répartition du courant à la surface de l'électrode interne. Le tube interne était alors rempli de poudre
de fer, qui se rétractait aux températures élevées et que nous avons remplacée, dans la suite, par de la poussière de graphite.
Les expériences 272 à 274 ont été effectuées avec du quartz nu; les expériences 275 à 278, en recouvrant le plus petit des tubes avec une couche mince de 50% d'oxyde de calcium et 50% d'oxyde de strontium. Les deux oxydes étaient mélangés avec de l'eau. On recouvrait le tube de cette pâte, que l'on calcinait ensuite au chalumeau.
FIG. 3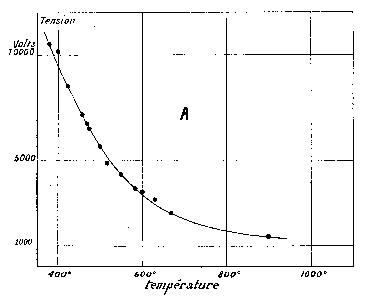
INTERPRÉTATION DES RÉSULTATS
La complexité des phénomènes qui se produisent dans l'effluve, impose une grande prudence dans l'interprétation des observations.
D'une façon générale, on ne saurait affirmer que la température ait une influence sur les rendements de l'effluve en oxydes d'azote au-dessous de 1000°. Il n'en est pas de même pour l'action du platine.
Ainsi qu'on peut s'en rendre compte par l'examen du tableau précédent, en comparant. des exépriences effectuées dans des conditions identiques de pression et de débit, les rendements, en présence du platine présentent des irrégularités telles qu'une courbe réunissant les points représentatifs de ces rendements en fonction dela température, n'aurait aucune signification. Par contre, ainsi qu'on le voit sur les courbes B et C de la figure 3, représentant les rendements rapportés à la durée de fonctionnement de l'effluveur (supposée proportionnelle au nombre d'expériences), l'activité du platine, après un certain temps dc préparation, passe par un maximum très net. Puis elle diminue rapidement et les rendements ne dépassent pas ceux que l'on obtient avec le quartz nu. Cette activité n'est pas liée à une diminution de la résistance électrique de l'effluve. Elle est bien due à l'activité catalytique du platine.
Avec le mélange d'oxydes, on remarque, par contre, une diminution sensible de la résistance de l'effluve mais les rendements ne sont pas modifiés. La diminution de résistance de l'effluve est due sans doute à l'émission électronique, mais, aux températures inférieures à 1000°, il ne se produit pas cette action favorable que l'on observe à 1600° 1.
RÉSUMè
1. Nous avons perfectionné la méthode d'analyse des oxydes d'azote par réduction en ammoniac, ce qui nous a permis d'évaluer 0,0008 gr. d'HNO 3 avec une précision de 1 %.
2. Nous avons pu montrer, grâce à cette méthode d'analyse, que l'ozone ne peut oxyder de lui-même l'azote, en quantité appréciable, comme l'avaient supposé certains auteurs, même à des concentrations très élevées.
3. Nous avons établi un effluveur à deux diélectriques, en quartz, permettant de. soumettre à l'effluve un mélange gazeux à des températures atteignant 1000° ou même davantage.
4. Nous avons étudié la formation des oxydes d'azote à partir de l'air atmosphérique, dans l'effluve, à des températures croissantes, jusqu'à 1000°. Dans ces limites, les rendements n'ont pas subi des modifications importantes.
5. Nous avons étudié l'action du platine et d'un mélange d' oxydes alcalino-terreux sur la formation de l'oxyde d'azote par l'effluve, à des températures comprises entre 600° et 1000°. L'action du platine, après un certain temps de préparation, présente un maximum très marqué, puis s'atténue peu à peu. Le mélange d'oxyde considéré n'a pas d'action sensible.

